Byetta for Treatment of Diabetes - Byetta, Full Prescribing Information
Brand Name: Byetta
Generic Name: Exenatide
Dosage Form: Injection
Contents:
Description
Clinical Pharmacology
Clinical Studies
Indications and Usage
Contraindications
Precautions
Adverse Reactions
Overdosage
Dosage and Administration
Storage
How Supplied
Byetta (Exenatide) Patient Information (in plain English)
Description
Byetta® (exenatide) is a synthetic peptide that has incretin-mimetic actions and was originally identified in the lizard Heloderma suspectum. Byetta enhances glucose-dependent insulin secretion by the pancreatic beta-cell, suppresses inappropriately elevated glucagon secretion, and slows gastric emptying. Exenatide differs in chemical structure and pharmacological action from insulin, sulfonylureas (including D-phenylalanine derivatives and meglitinides), biguanides, thiazolidinediones, and alpha-glucosidase inhibitors.
Exenatide is a 39−amino acid peptide amide. Exenatide has the empirical formula C184H282N50O60S and molecular weight of 4186.6 Daltons. The amino acid sequence for exenatide is shown below.
H - His - Gly - Glu - Gly - Thr - Phe - Thr - Ser - Asp - Leu - Ser - Lys - Gln - Met - Glu - Glu - Glu - Ala - Val - Arg - Leu - Phe - Ile - Glu - Trp - Leu - Lys - Asn - Gly - Gly - Pro - Ser - Ser - Gly - Ala - Pro - Pro - Pro - Ser - NH2
Byetta is supplied for subcutaneous (SC) injection as a sterile, preserved isotonic solution in a glass cartridge that has been assembled in a pen-injector (pen). Each milliliter (mL) contains 250 micrograms (mcg) synthetic exenatide, 2.2 mg metacresol as an antimicrobial preservative, mannitol as a tonicity-adjusting agent, and glacial acetic acid and sodium acetate trihydrate in water for injection as a buffering solution at pH 4.5. Two prefilled pens are available to deliver unit doses of 5 mcg or 10 mcg. Each prefilled pen will deliver 60 doses to provide 30 days of twice daily administration (BID).
Clinical Pharmacology
Mechanism of Action
Incretins, such as glucagon-like peptide-1 (GLP-1), enhance glucose-dependent insulin secretion and exhibit other antihyperglycemic actions following their release into the circulation from the gut. Exenatide is an incretin mimetic agent that mimics the enhancement of glucose-dependent insulin secretion and several other antihyperglycemic actions of incretins.
The amino acid sequence of exenatide partially overlaps that of human GLP-1. Exenatide has been shown to bind and activate the known human GLP-1 receptor in vitro. This leads to an increase in both glucose-dependent synthesis of insulin, and in vivo secretion of insulin from pancreatic beta cells, by mechanisms involving cyclic AMP and/or other intracellular signaling pathways. Exenatide promotes insulin release from beta cells in the presence of elevated glucose concentrations. When administered in vivo, exenatide mimics certain antihyperglycemic actions of GLP-1.
Byetta improves glycemic control by reducing fasting and postprandial glucose concentrations in patients with type 2 diabetes through the actions described below.
Glucose-dependent insulin secretion: Byetta has acute effects on pancreatic beta-cell responsiveness to glucose and leads to insulin release only in the presence of elevated glucose concentrations. This insulin secretion subsides as blood glucose concentrations decrease and approach euglycemia.
First-phase insulin response: In healthy individuals, robust insulin secretion occurs during the first 10 minutes following intravenous (IV) glucose administration. This secretion, known as the "first-phase insulin response," is characteristically absent in patients with type 2 diabetes. The loss of the first-phase insulin response is an early beta-cell defect in type 2 diabetes. Administration of Byetta at therapeutic plasma concentrations restored first-phase insulin response to an IV bolus of glucose in patients with type 2 diabetes (Figure 1). Both first-phase insulin secretion and second-phase insulin secretion were significantly increased in patients with type 2 diabetes treated with Byetta compared with saline (p
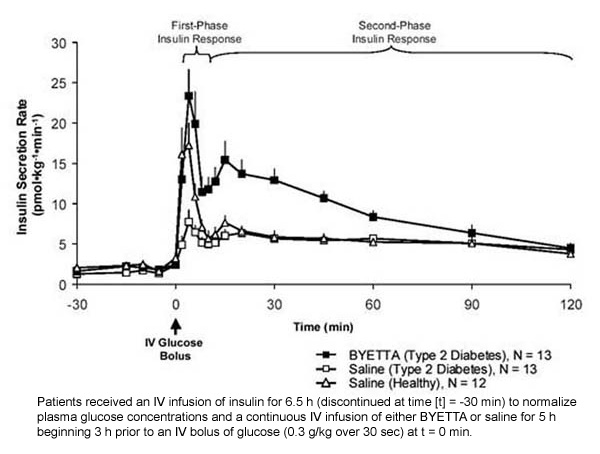
Figure 1: Mean (+SEM) Insulin Secretion Rate During Infusion of Byetta or Saline in Patients With Type 2 Diabetes and During Infusion of Saline in Healthy Subjects
Glucagon secretion: In patients with type 2 diabetes, Byetta moderates glucagon secretion and lowers serum glucagon concentrations during periods of hyperglycemia. Lower glucagon concentrations lead to decreased hepatic glucose output and decreased insulin demand. However, Byetta does not impair the normal glucagon response to hypoglycemia.
Gastric emptying: Byetta slows gastric emptying, thereby reducing the rate at which meal-derived glucose appears in the circulation.
Food intake: In both animals and humans, administration of exenatide has been shown to reduce food intake.
Pharmacokinetics
Absorption
Following SC administration to patients with type 2 diabetes, exenatide reaches median peak plasma concentrations in 2.1 h. Mean peak exenatide concentration (Cmax) was 211 pg/mL and overall mean area under the curve (AUC0-inf) was 1036 pg-h/mL following SC administration of a 10 mcg dose of Byetta. Exenatide exposure (AUC) increased proportionally over the therapeutic dose range of 5 mcg to 10 mcg. The Cmax values increased less than proportionally over the same range. Similar exposure is achieved with SC administration of Byetta in the abdomen, thigh, or arm.
Distribution
The mean apparent volume of distribution of exenatide following SC administration of a single dose of Byetta is 28.3 L.
Metabolism and Elimination
Nonclinical studies have shown that exenatide is predominantly eliminated by glomerular filtration with subsequent proteolytic degradation. The mean apparent clearance of exenatide in humans is 9.1 L/h and the mean terminal half-life is 2.4 h. These pharmacokinetic characteristics of exenatide are independent of the dose. In most individuals, exenatide concentrations are measurable for approximately 10 h post-dose.
Special Populations
Renal Insufficiency
In patients with mild to moderate renal impairment (creatinine clearance 30 to 80 mL/min), exenatide clearance was only mildly reduced; therefore, no dosage adjustment of Byetta is required in patients with mild to moderate renal impairment. However, in patients with end-stage renal disease receiving dialysis, mean exenatide clearance is reduced to 0.9 L/h compared with 9.1 L/h in healthy subjects (see PRECAUTIONS, General).
Hepatic Insufficiency
No pharmacokinetic study has been performed in patients with a diagnosis of acute or chronic hepatic insufficiency. Because exenatide is cleared primarily by the kidney, hepatic dysfunction is not expected to affect blood concentrations of exenatide (see Pharmacokinetics, Metabolism and Elimination).
Geriatric
Population pharmacokinetic analysis of patients (range from 22 to 73 years) suggests that age does not influence the pharmacokinetic properties of exenatide.
Pediatric
Exenatide has not been studied in pediatric patients.
Gender
Population pharmacokinetic analysis of male and female patients suggests that gender does not influence the distribution and elimination of exenatide.
Race
Population pharmacokinetic analysis of patients including Caucasian, Hispanic, and Black, suggests that race has no significant influence on the pharmacokinetics of exenatide.
Obesity
Population pharmacokinetic analysis of obese (BMI ≥30 kg/m2) and non-obese patients suggests that obesity has no significant effect on the pharmacokinetics of exenatide.
Drug Interactions
Digoxin
Coadministration of repeated doses of Byetta (10 mcg BID) decreased the Cmax of oral digoxin (0.25 mg QD) by 17% and delayed the Tmax by approximately 2.5 h; however, the overall steady-state pharmacokinetic exposure (AUC) was not changed.
Lovastatin
Lovastatin AUC and Cmax were decreased approximately 40% and 28%, respectively, and Tmax was delayed about 4 h when Byetta (10 mcg BID) was administered concomitantly with a single dose of lovastatin (40 mg) compared with lovastatin administered alone. In the 30-week controlled clinical trials of Byetta, the use of Byetta in patients already receiving HMG CoA reductase inhibitors was not associated with consistent changes in lipid profiles compared to baseline.
Lisinopril
In patients with mild to moderate hypertension stabilized on lisinopril (5 to 20 mg/day), Byetta (10 mcg BID) did not alter steady-state Cmax or AUC of lisinopril. Lisinopril steady-state Tmax was delayed by 2 h. There were no changes in 24-h mean systolic and diastolic blood pressure.
Acetaminophen
When 1000 mg acetaminophen elixir was given with 10 mcg Byetta (0 h) and 1 h, 2 h, and 4 h after Byetta injection, acetaminophen AUCs were decreased by 21%, 23%, 24%, and 14%, respectively; Cmax was decreased by 37%, 56%, 54%, and 41%, respectively; Tmax was increased from 0.6 h in the control period to 0.9 h, 4.2 h, 3.3 h, and 1.6 h, respectively. Acetaminophen AUC, Cmax and Tmax were not significantly changed when acetaminophen was given 1 h before Byetta injection.
Warfarin
Coadministration of repeat doses of Byetta (5 mcg BID on days 1-2 and 10 mcg BID on days 3-9) in healthy volunteers, delayed warfarin (25 mg) Tmax by about 2 h. No clinically relevant effects on Cmax or AUC of S- and R-enantiomers of warfarin were observed. Byetta did not change the pharmacodynamic properties (as assessed by INR response) of warfarin.
Pharmacodynamics
Postprandial Glucose
In patients with type 2 diabetes, Byetta reduces the postprandial plasma glucose concentrations (Figure 2).
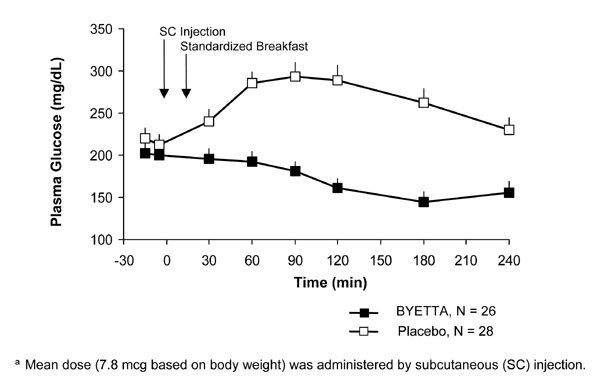
Figure 2: Mean (+SEM) Postprandial Plasma Glucose Concentrations on Day 1 of Byettaa Treatment in Patients With Type 2 Diabetes Treated With Metformin, a Sulfonylurea, or Both (N = 54)
Fasting Glucose
In a single-dose crossover study in patients with type 2 diabetes and fasting hyperglycemia, an immediate insulin release followed injection of Byetta. Plasma glucose concentrations were significantly reduced with Byetta compared with placebo (Figure 3).
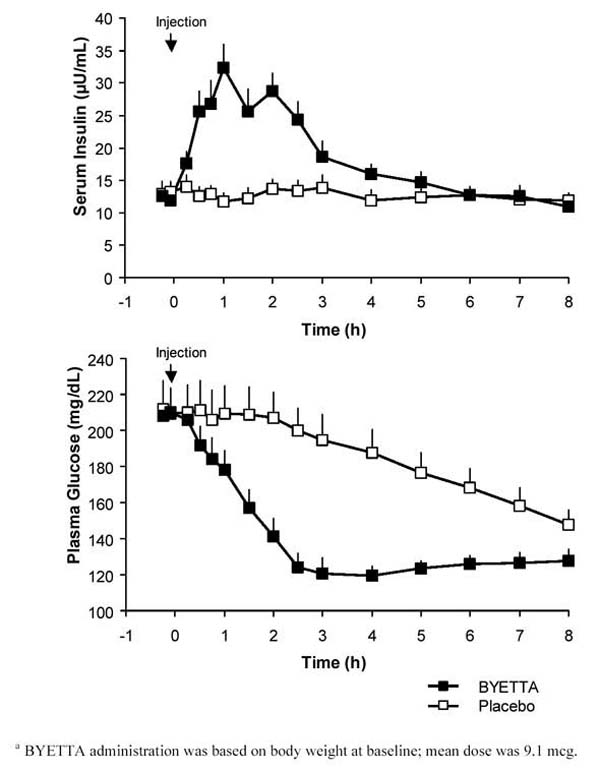
Figure 3: Mean (+SEM) Serum Insulin and Plasma Glucose Concentrations Following a One-Time Injection of Byettaa or Placebo in Fasting Patients With Type 2 Diabetes (N = 12)
Clinical Studies
Use with metformin and/or a sulfonylurea
Three 30-week, double-blind, placebo-controlled trials were conducted to evaluate the safety and efficacy of Byetta in patients with type 2 diabetes whose glycemic control was inadequate with metformin alone, a sulfonylurea alone, or metformin in combination with a sulfonylurea.
A total of 1446 patients were randomized in these three trials: 991 (68.5%) were Caucasian, 224 (15.5%) were Hispanic, and 174 (12.0%) were Black. Mean HbA1c values at baseline for the trials ranged from 8.2% to 8.7%. After a 4-week placebo lead-in period, patients were randomly assigned to receive Byetta 5 mcg BID, Byetta 10 mcg BID, or placebo BID before the morning and evening meals, in addition to their existing oral antidiabetic agent. All patients assigned to Byetta began a treatment initiation period with 5 mcg BID for 4 weeks. After 4 weeks, those patients either continued to receive Byetta 5 mcg BID or had their dose increased to 10 mcg BID. Patients assigned to placebo received placebo BID throughout the study.
The primary endpoint in each study was mean change from baseline HbA1c at 30 weeks. Thirty-week study results are summarized in Table 1.
Table 1: Results of Thirty-Week Placebo-Controlled Trials of Byetta in Patients With Inadequate Glucose Control Despite the Use of Metformin, a Sulfonylurea, or Both
| Placebo BID | Byetta 5 mcg BID | Byetta 10 mcg* BID | |
|---|---|---|---|
| |||
| In Combination With Metformin | |||
| Intent-to-Treat Population (N) | 113 | 110 | 113 |
| HbA1c (%), Mean | |||
| Baseline | 8.2 | 8.3 | 8.2 |
| Change at Week 30 | +0.1 | −0.4†| −0.8c |
| Proportion Achieving HbA1c≤7% § | 13.0% | 31.6%†| 46.4%†|
| Body Weight (kg), Mean | |||
| Baseline | 99.9 | 100.0 | 100.9 |
| Change at Week 30 | −0.3 | −1.6†| −2.8c |
| In Combination With a Sulfonylurea | |||
| Intent-to-Treat Population (N) | 123 | 125 | 129 |
| HbA1c (%), Mean | |||
| Baseline | 8.7 | 8.5 | 8.6 |
| Change at Week 30 | +0.1 | −0.5†| −0.9c |
| Proportion Achieving HbA1c≤7% § | 8.8% | 32.6%†| 41.3%c |
| Body Weight (kg), Mean | |||
| Baseline | 99.1 | 94.9 | 95.2 |
| Change at Week 30 | −0.6 | −0.9 | −1.6†|
| In Combination With Metformin and a Sulfonylurea | |||
| Intent-to-Treat Population (N) | 247 | 245 | 241 |
| HbA1c (%), Mean | |||
| Baseline | 8.5 | 8.5 | 8.5 |
| Change at Week 30 | +0.2 | −0.6c | −0.8c |
| Proportion Achieving HbA1c≤7% § | 9.2% | 27.4%c | 33.5%c |
| Body Weight (kg), Mean | |||
| Baseline | 99.1 | 96.9 | 98.4 |
| Change at Week 30 | −0.9 | −1.6†| −1.6†|
HbA1c
The addition of Byetta to a regimen of metformin, a sulfonylurea, or both, resulted in statistically significant reductions from baseline HbA1c at Week 30 compared with patients receiving placebo added to these agents in the three controlled trials (Table 1). In addition, a statistically significant dose-effect was observed between 5-mcg and 10-mcg Byetta groups for the change from baseline HbA1c at Week 30 in the three studies.
Fasting and Postprandial Glucose
Long-term use of Byetta in combination with metformin, a sulfonylurea, or both, reduced both fasting and postprandial plasma glucose concentrations in a statistically significant, dose-dependent manner through Week 30. A statistically significant reduction from baseline in both mean fasting and postprandial glucose concentrations was observed at Week 30 in both Byetta groups compared with placebo in data combined from the three controlled trials. The change in fasting glucose concentration at Week 30 compared with baseline was −8 mg/dL for Byetta 5 mcg BID and −10 mg/dL for Byetta 10 mcg BID, compared with +12 mg/dL for placebo. The change in 2-h postprandial glucose concentration following administration of Byetta at Week 30 compared with baseline was −63 mg/dL for 5 mcg BID and −71 mg/dL for 10 mcg BID, compared with +11 mg/dL for placebo.
Proportion of Patients Achieving HbA1c≤7%
Byetta in combination with metformin, a sulfonylurea, or both, resulted in a greater, statistically significant proportion of patients achieving an HbA1c≤7% at Week 30 compared with patients receiving placebo in combination with these agents (Table 1).
Body Weight
In the three controlled trials, a decrease from baseline body weight at Week 30 was associated with Byetta 10 mcg BID compared with placebo BID in patients with type 2 diabetes (Table 1).
One-Year Clinical Results
The cohort of 163 patients from the 30-week placebo-controlled trials who completed a total of 52 weeks of treatment with Byetta 10 mcg BID had HbA1c changes from baseline of −1.0% and −1.1% at 30 and 52 weeks of treatment, respectively, with accompanying changes from baseline in fasting plasma glucose of −14.0 mg/dL and −25.3 mg/dL, and body weight changes of −2.6 kg and −3.6 kg. This cohort had baseline values similar to those of the entire controlled-trial population.
Use with a thiazolidinedione
In a randomized, double-blind, placebo-controlled trial of 16 weeks duration, Byetta (n=121) or placebo (n=112) was added to existing thiazolidinedione (pioglitazone or rosiglitazone) treatment, with or without metformin, in patients with type 2 diabetes with inadequate glycemic control. Randomization to Byetta or placebo was stratified based on whether the patients were receiving metformin. Patients assigned to placebo received placebo BID throughout the study. Byetta or placebo was injected subcutaneously before the morning and evening meals. Seventy-nine percent of patients were taking a thiazolidinedione and metformin and 21% were taking a thiazolidinedione alone. The majority of patients (84%) were Caucasian, 8% were Hispanic and 3% were Black. The mean baseline HbA1c values were similar for Byetta and placebo (7.9%). Byetta treatment was initiated at a dose of 5 mcg BID for 4 weeks then increased to 10 mcg BID for 12 more weeks.
Sixteen-week study results are summarized in Table 2. Compared to placebo, Byetta resulted in statistically significant reductions in HbA1c from baseline at Week 16. Treatment effects for HbA1c were similar in the two sub-groups defined by underlying treatment stratum (thiazolidinediones alone versus thiazolidinediones plus metformin). The change in fasting serum glucose concentration from baseline to Week 16 was statistically significant compared with placebo (−21 mg/dL for Byetta 10 mcg BID compared with +4 mg/dL for placebo).
Table 2: Results of 16-Week Placebo-Controlled Trial of Byetta in Patients With Inadequate Glucose Control Despite the Use of a Thiazolidinedione (TZD) or a Thiazolidinedione plus Metformin
| Placebo BID | Byetta 10 mcg* BID | ||
|---|---|---|---|
| |||
| In Combination With a TZD or a TZD plus MET | |||
| Intent-to-Treat Population (N) | 112 | 121 | |
| HbA1c (%), Mean | |||
| Baseline | 7.9 | 7.9 | |
| Change at Week 16 | +0.1 | −0.8†| |
| Proportion Achieving HbA1c≤7%c | 16.2% | 62.3%†| |
| Body Weight (kg), Mean | |||
| Baseline | 96.9 | 97.5 | |
| Change at Week 16 | −0.2 | −1.5†| |
Indications and Usage
Byetta is indicated as adjunctive therapy to improve glycemic control in patients with type 2 diabetes mellitus who are taking metformin, a sulfonylurea, a thiazolidinedione, a combination of metformin and a sulfonylurea, or a combination of metformin and a thiazolidinedione, but have not achieved adequate glycemic control.
Contraindications
Byetta is contraindicated in patients with known hypersensitivity to exenatide or to any of the product components.
Precautions
General
Byetta is not a substitute for insulin in insulin-requiring patients. Byetta should not be used in patients with type 1 diabetes or for the treatment of diabetic ketoacidosis.
Patients may develop anti-exenatide antibodies following treatment with Byetta, consistent with the potentially immunogenic properties of protein and peptide pharmaceuticals. Patients receiving Byetta should be observed for signs and symptoms of hypersensitivity reactions.
In a small proportion of patients, the formation of anti-exenatide antibodies at high titers could result in failure to achieve adequate improvement in glycemic control. If there is worsening glycemic control or failure to achieve targeted glycemic control, alternative antidiabetic therapy should be considered.
The concurrent use of Byetta with insulin, D-phenylalanine derivatives, meglitinides, or alpha-glucosidase inhibitors has not been studied.
Byetta is not recommended for use in patients with end-stage renal disease or severe renal impairment (creatinine clearance Pharmacokinetics, Special Populations). In patients with end-stage renal disease receiving dialysis, single doses of Byetta 5 mcg were not well tolerated due to gastrointestinal side effects.
There have been rare, spontaneously reported events of altered renal function, including increased serum creatinine, renal impairment, worsened chronic renal failure and acute renal failure, sometimes requiring hemodialysis. Some of these events occurred in patients receiving one or more pharmacologic agents known to affect renal function/hydration status and/or in patients experiencing nausea, vomiting, and/or diarrhea, with or without dehydration. Concomitant agents included angiotensin converting enzyme inhibitors, nonsteroidal anti-inflammatory drugs, and diuretics. Reversibility of altered renal function has been observed with supportive treatment and discontinuation of potentially causative agents, including exenatide. Exenatide has not been found to be directly nephrotoxic in preclinical or clinical studies.
Byetta has not been studied in patients with severe gastrointestinal disease, including gastroparesis. Its use is commonly associated with gastrointestinal adverse effects, including nausea, vomiting, and diarrhea. Therefore, the use of Byetta is not recommended in patients with severe gastrointestinal disease. The development of severe abdominal pain in a patient treated with Byetta should be investigated because it may be a warning sign of a serious condition.
Hypoglycemia
In the 30-week controlled clinical trials with Byetta, a hypoglycemia episode was recorded as an adverse event if the patient reported symptoms associated with hypoglycemia with an accompanying blood glucose DOSAGE AND ADMINISTRATION).
Table 3: Incidence (%) of Hypoglycemia* by Concomitant Antidiabetic Therapy
| Byetta | Byetta | Byetta | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Placebo BID | 5 mcg BID | 10 mcg BID | Placebo BID | 5 mcg BID | 10 mcg BID | Placebo BID | 5 mcg BID | 10 mcg BID | |
| With Metformin | With a Sulfonylurea | With MET/SFU | |||||||
| Byetta and placebo were administered before the morning and evening meals. | |||||||||
| Abbreviations: BID, twice daily; MET/SFU, metformin and a sulfonylurea. | |||||||||
| |||||||||
| N | 113 | 110 | 113 | 123 | 125 | 129 | 247 | 245 | 241 |
| Hypoglycemia | 5.3% | 4.5% | 5.3% | 3.3% | 14.4% | 35.7% | 12.6% | 19.2% | 27.8% |
When used as add-on to a thiazolidinedione, with or without metformin, the incidence of symptomatic mild to moderate hypoglycemia with Byetta was 11% compared to 7% with placebo.
Byetta did not alter the counter-regulatory hormone responses to insulin-induced hypoglycemia in a randomized, double-blind, controlled study in healthy subjects.
Information for Patients
Patients should be informed of the potential risks of Byetta. Patients should also be fully informed about self-management practices, including the importance of proper storage of Byetta, injection technique, timing of dosage of Byetta as well as concomitant oral drugs, adherence to meal planning, regular physical activity, periodic blood glucose monitoring and HbA1c testing, recognition and management of hypoglycemia and hyperglycemia, and assessment for diabetes complications.
Patients should be advised to inform their physicians if they are pregnant or intend to become pregnant.
Each dose of Byetta should be administered as a SC injection in the thigh, abdomen, or upper arm at any time within the 60-minute period before the morning and evening meals (or before the two main meals of the day, approximately 6 hours or more apart). Byetta should not be administered after a meal. If a dose is missed, the treatment regimen should be resumed as prescribed with the next scheduled dose.
The risk of hypoglycemia is increased when Byetta is used in combination with an agent that induces hypoglycemia, such as a sulfonylurea. The symptoms, treatment, and conditions that predispose development of hypoglycemia should be explained to the patient. While the patient's usual instructions for hypoglycemia management do not need to be changed, these instructions should be reviewed and reinforced when initiating Byetta therapy, particularly when concomitantly administered with a sulfonylurea (see PRECAUTIONS, Hypoglycemia).
Patients should be advised that treatment with Byetta may result in a reduction in appetite, food intake, and/or body weight, and that there is no need to modify the dosing regimen due to such effects. Treatment with Byetta may also result in nausea, particularly upon initiation of therapy (see ADVERSE REACTIONS).
The patient should read the "Information for the Patient" insert and the Pen User Manual before starting Byetta therapy and review them each time the prescription is refilled. The patient should be instructed on proper use and storage of the pen, emphasizing how and when to set up a new pen and noting that only one setup step is necessary at initial use. The patient should be advised not to share the pen and needles.
Patients should be informed that pen needles are not included with the pen and must be purchased separately. Patients should be advised which needle length and gauge should be used.
Drug Interactions
The effect of Byetta to slow gastric emptying may reduce the extent and rate of absorption of orally administered drugs. Byetta should be used with caution in patients receiving oral medications that require rapid gastrointestinal absorption. For oral medications that are dependent on threshold concentrations for efficacy, such as contraceptives and antibiotics, patients should be advised to take those drugs at least 1 h before Byetta injection. If such drugs are to be administered with food, patients should be advised to take them with a meal or snack when Byetta is not administered. The effect of Byetta on the absorption and effectiveness of oral contraceptives has not been characterized.
Warfarin
In a controlled clinical pharmacology study in healthy volunteers, a delay in warfarin Tmax of about 2 h was observed when warfarin was administered 30 min after Byetta. No clinically relevant effects on Cmax or AUC were observed. However, since market introduction there have been some spontaneously reported cases of increased INR (International Normalized Ratio) with concomitant use of warfarin and Byetta, sometimes associated with bleeding.
Carcinogenesis, Mutagenesis, Impairment of Fertility
A 104-week carcinogenicity study was conducted in male and female rats at doses of 18, 70, or 250 mcg/kg/day administered by bolus SC injection. Benign thyroid C-cell adenomas were observed in female rats at all exenatide doses. The incidences in female rats were 8% and 5% in the two control groups and 14%, 11%, and 23% in the low-, medium-, and high-dose groups with systemic exposures of 5, 22, and 130 times, respectively, the human exposure resulting from the maximum recommended dose of 20 mcg/day, based on plasma area under the curve (AUC).
In a 104-week carcinogenicity study in mice at doses of 18, 70, or 250 mcg/kg/day administered by bolus SC injection, no evidence of tumors was observed at doses up to 250 mcg/kg/day, a systemic exposure up to 95 times the human exposure resulting from the maximum recommended dose of 20 mcg/day, based on AUC.
Exenatide was not mutagenic or clastogenic, with or without metabolic activation, in the Ames bacterial mutagenicity assay or chromosomal aberration assay in Chinese hamster ovary cells. Exenatide was negative in the in vivo mouse micronucleus assay.
In mouse fertility studies with SC doses of 6, 68 or 760 mcg/kg/day, males were treated for 4 weeks prior to and throughout mating and females were treated 2 weeks prior to and throughout mating until gestation day 7. No adverse effect on fertility was observed at 760 mcg/kg/day, a systemic exposure 390 times the human exposure resulting from the maximum recommended dose of 20 mcg/day, based on AUC.
Pregnancy
Pregnancy Category C
Exenatide has been shown to cause reduced fetal and neonatal growth, and skeletal effects in mice at systemic exposures 3 times the human exposure resulting from the maximum recommended dose of 20 mcg/day, based on AUC. Exenatide has been shown to cause skeletal effects in rabbits at systemic exposures 12 times the human exposure resulting from the maximum recommended dose of 20 mcg/day, based on AUC. There are no adequate and well-controlled studies in pregnant women. Byetta should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus.
In female mice given SC doses of 6, 68, or 760 mcg/kg/day beginning 2 weeks prior to and throughout mating until gestation day 7, there were no adverse fetal effects at doses up to 760 mcg/kg/day, systemic exposures up to 390 times the human exposure resulting from the maximum recommended dose of 20 mcg/day, based on AUC.
In pregnant mice given SC doses of 6, 68, 460, or 760 mcg/kg/day from gestation day 6 through 15 (organogenesis), cleft palate (some with holes) and irregular skeletal ossification of rib and skull bones were observed at 6 mcg/kg/day, a systemic exposure 3 times the human exposure resulting from the maximum recommended dose of 20 mcg/kg/day, based on AUC.
In pregnant rabbits given SC doses of 0.2, 2, 22, 156, or 260 mcg/kg/day from gestation day 6 through 18 (organogenesis), irregular skeletal ossifications were observed at 2 mcg/kg/day, a systemic exposure 12 times the human exposure resulting from the maximum recommended dose of 20 mcg/day, based on AUC.
In pregnant mice given SC doses of 6, 68, or 760 mcg/kg/day from gestation day 6 through lactation day 20 (weaning), an increased number of neonatal deaths were observed on postpartum days 2-4 in dams given 6 mcg/kg/day, a systemic exposure 3 times the human exposure resulting from the maximum recommended dose of 20 mcg/day, based on AUC.
Nursing Mothers
It is not known whether exenatide is excreted in human milk. Many drugs are excreted in human milk and because of the potential for clinically significant adverse reactions in nursing infants from exenatide, a decision should be made whether to discontinue producing milk for consumption or discontinue the drug, taking into account the importance of the drug to the lactating woman. Studies in lactating mice have demonstrated that exenatide is present at low concentrations in milk (less than or equal to 2.5% of the concentration in maternal plasma following subcutaneous dosing). Caution should be exercised when Byetta is administered to a nursing woman.
Pediatric Use
Safety and effectiveness of Byetta have not been established in pediatric patients.
Geriatric Use
Byetta was studied in 282 patients 65 years of age or older and in 16 patients 75 years of age or older. No differences in safety or effectiveness were observed between these patients and younger patients.
Adverse Reactions
Use with metformin and/or a sulfonylurea
In the three 30-week controlled trials of Byetta add-on to metformin and/or sulfonylurea, adverse events with an incidence ≥5% (excluding hypoglycemia; see Table 3) that occurred more frequently in Byetta-treated patients compared with placebo-treated patients are summarized in Table 4.
Table 4: Frequent Treatment-Emergent Adverse Events (≥5% Incidence and Greater Incidence With Byetta Treatment) Excluding Hypoglycemia*
| Placebo BID N = 483 % | All Byetta BID N = 963 % | |
|---|---|---|
| ||
| Nausea | 18 | 44 |
| Vomiting | 4 | 13 |
| Diarrhea | 6 | 13 |
| Feeling Jittery | 4 | 9 |
| Dizziness | 6 | 9 |
| Headache | 6 | 9 |
| Dyspepsia | 3 | 6 |
The adverse events associated with Byetta generally were mild to moderate in intensity. The most frequently reported adverse event, mild to moderate nausea, occurred in a dose-dependent fashion. With continued therapy, the frequency and severity decreased over time in most of the patients who initially experienced nausea. Adverse events reported in ≥1.0 to <5.0% of patients receiving Byetta and reported more frequently than with placebo included asthenia (mostly reported as weakness), decreased appetite, gastroesophageal reflux disease, and hyperhidrosis. Patients in the extension studies at 52 weeks experienced similar types of adverse events observed in the 30-week controlled trials.
The incidence of withdrawal due to adverse events was 7% for Byetta-treated patients and 3% for placebo-treated patients. The most common adverse events leading to withdrawal for Byetta-treated patients were nausea (3% of patients) and vomiting (1%). For placebo-treated patients, <1% withdrew due to nausea and 0% due to vomiting.
Use with a thiazolidinedione
In the 16-week placebo-controlled study of Byetta add-on to a thiazolidinedione, with or without metformin, the incidence and type of other adverse events observed were similar to those seen in the 30-week controlled clinical trials with metformin and/or a sulfonylurea. No serious adverse events were reported in the placebo arm. Two serious adverse events, namely chest pain (leading to withdrawal) and chronic hypersensitivity pneumonitis, were reported in the Byetta arm.
The incidence of withdrawal due to adverse events was 16% (19/121) for Byetta-treated patients and 2% (2/112) for placebo-treated patients. The most common adverse events leading to withdrawal for Byetta-treated patients were nausea (9%) and vomiting (5%). For placebo-treated patients, <1% withdrew due to nausea. Chills (n=4) and injection-site reactions (n=2) occurred only in Byetta-treated patients. The two patients who reported an injection-site reaction had high titers of anti-exenatide antibody.
Spontaneous Data
Since market introduction of Byetta, the following additional adverse reactions have been reported. Because these events are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
General: injection-site reactions; dysgeusia; somnolence, INR increased with concomitant warfarin use (some reports associated with bleeding).
Allergy/Hypersensitivity: generalized pruritus and/or urticaria, macular or papular rash, angioedema; rare reports of anaphylactic reaction.
Gastrointestinal: nausea, vomiting, and/or diarrhea resulting in dehydration; abdominal distension, abdominal pain, eructation, constipation, flatulence, acute pancreatitis.
Renal and Urinary Disorders: altered renal function, including acute renal failure, worsened chronic renal failure, renal impairment, increased serum creatinine (see PRECAUTIONS).
Immunogenicity
Consistent with the potentially immunogenic properties of protein and peptide pharmaceuticals, patients may develop anti-exenatide antibodies following treatment with Byetta. In most patients who develop antibodies, antibody titers diminish over time.
In the 30-week controlled trials of Byetta add-on to metformin and/or sulfonylurea, 38% of patients had low titer anti-exenatide antibodies at 30 weeks. For this group, the level of glycemic control (HbA1c) was generally comparable to that observed in those without antibody titers. An additional 6% of patients had higher titer antibodies at 30 weeks. In about half of this 6% (3% of the total patients given Byetta in the 30-week controlled studies), the glycemic response to Byetta was attenuated; the remainder had a glycemic response comparable to that of patients without antibodies.
In the 16-week trial of Byetta add-on to thiazolidinediones, with or without metformin, 9% of patients had higher titer antibodies at 16 weeks. Compared with patients who did not develop antibodies to Byetta, on average the glycemic response in patients with higher titer antibodies was attenuated.
The patient's glycemic response to Byetta should be monitored. If there is worsening glycemic control or failure to achieve targeted glycemic control, alternative antidiabetic therapy should be considered.
Overdosage
In a clinical study of Byetta, three patients with type 2 diabetes each experienced a single overdose of 100 mcg SC (10 times the maximum recommended dose). Effects of the overdoses included severe nausea, severe vomiting, and rapidly declining blood glucose concentrations. One of the three patients experienced severe hypoglycemia requiring parenteral glucose administration. The three patients recovered without complication. In the event of overdose, appropriate supportive treatment should be initiated according to the patient's clinical signs and symptoms.
Dosage and Administration
Byetta therapy should be initiated at 5 mcg per dose administered twice daily at any time within the 60-minute period before the morning and evening meals (or before the two main meals of the day, approximately 6 hours or more apart). Byetta should not be administered after a meal. Based on clinical response, the dose of Byetta can be increased to 10 mcg twice daily after 1 month of therapy. Each dose should be administered as a SC injection in the thigh, abdomen, or upper arm.
Byetta is recommended for use in patients with type 2 diabetes mellitus who are already receiving metformin, a sulfonylurea, a thiazolidinedione, a combination of metformin and a sulfonylurea, or a combination of metformin and a thiazolidinedione, and have suboptimal glycemic control. When Byetta is added to metformin or thiazolidinedione therapy, the current dose of metformin or thiazolidinedione can be continued as it is unlikely that the dose of metformin or thiazolidinedione will require adjustment due to hypoglycemia when used with Byetta. When Byetta is added to sulfonylurea therapy, a reduction in the dose of sulfonylurea may be considered to reduce the risk of hypoglycemia (see PRECAUTIONS, Hypoglycemia).
Byetta is a clear and colorless liquid and should not be used if particles appear or if the solution is cloudy or colored. Byetta should not be used past the expiration date. No data are available on the safety or efficacy of intravenous or intramuscular injection of Byetta.
Storage
Prior to first use, Byetta must be stored refrigerated at 36°F to 46°F (2°C to 8°C). After first use, Byetta can be kept at a temperature not to exceed 77°F (25°C). Do not freeze. Do not use Byetta if it has been frozen. Byetta should be protected from light. The pen should be discarded 30 days after first use, even if some drug remains in the pen.
How is Supplied
Byetta is supplied as a sterile solution for subcutaneous injection containing 250 mcg/mL exenatide. The following packages are available:
5 mcg per dose, 60 doses, 1.2 mL prefilled pen NDC 66780-210-07
10 mcg per dose, 60 doses, 2.4 mL prefilled pen NDC 66780-210-08
Rx ONLY
Manufactured for Amylin Pharmaceuticals, Inc., San Diego, CA 92121
Marketed by Amylin Pharmaceuticals, Inc. and Eli Lilly and Company
1-800-868-1190
http://www.Byetta.com
Byetta is a registered trademark of Amylin Pharmaceuticals, Inc.
© 2007 Amylin Pharmaceuticals, Inc. All rights reserved.
last updated 09/2007
Byetta (Exenatide) Patient Information (in plain English)
Detailed Info on Signs, Symptoms, Causes, Treatments of Diabetes
The information in this monograph is not intended to cover all possible uses, directions, precautions, drug interactions or adverse effects. This information is generalized and is not intended as specific medical advice. If you have questions about the medicines you are taking or would like more information, check with your doctor, pharmacist, or nurse.
back to: Browse all Medications for Diabetes
APA Reference
Staff, H.
(2007, September 28). Byetta for Treatment of Diabetes - Byetta, Full Prescribing Information, HealthyPlace. Retrieved
on 2026, August 5 from https://www.healthyplace.com/diabetes/medications/byetta-diabetes-2-treatment